

La OI

¿Qué es la osteogénesis imperfecta?
La OI es una enfermedad congénita que se caracteriza por una formación inadecuada del tejido óseo. Los huesos de los afectados son muy frágiles y se fracturan con más facilidad. Existen otras manifestaciones, como baja estatura, problemas respiratorios, musculatura débil, pérdida de audición, problemas de visión, escoliosis y dolor crónico. Se considera una patología poco frecuente debido a que su prevalencia se estima entre 1 de cada 15.000 y 1 de cada 20.000 nacidos y se calcula que en España existen alrededor de 2700 afectados.
Si quieres saber más, visita nuestra biblioteca y descárgate nuestros boletines informativos.
¿Existe tratamiento?
Actualmente no hay cura para la OI. No obstante, existen tratamientos que ayudan a mejorar la calidad de vida de las personas afectadas:
A continuación puedes encontrar información mas detallada sobre la OI:
La osteogénesis imperfecta o “enfermedad de los huesos de cristal” es un trastorno genético congénito que comprende un alto espectro de presentaciones fenotípicas. Se trata de una patología del tejido conectivo que afecta a la producción del colágeno tipo 1, cuya función principal es proporcionar soporte y resistencia a la tracción de los tejidos. La consecuencia principal es la elevada fragilidad ósea, que puede dar lugar a fracturas frecuentes. Otras manifestaciones clínicas incluyen:
Deformidades y dolor óseo
Baja estatura
Escoliosis
Baja densidad ósea
Debilidad muscular, hiperlaxitud y esguinces
Pérdida de audición y problemas visuales
Dentinogénesis imperfecta
Problemas respiratorios, que se agravan con la presencia de deformidades del tórax y escoliosis
Complicaciones cardíacas
Fatiga
En los casos más severos, impresión basilar, que puede dar lugar a complicaciones neurológicas
Se considera una “enfermedad rara” o de baja prevalencia, debido a que se estima que afecta a 1 de cada 15.000/20.000 nacidos. Se calcula que en España existen unos 2700 afectados.
La clasificación de la OI está en constante cambio, como consecuencia de la investigación y los nuevos descrubrimientos. A continuación se presenta una clasificación muy completa y actualizada a 2024. La tabla está tomada de la publicación “Update on the Genetics of Osteogenesis Imperfecta”, de Jovanovic y Marini (2024):
‘
El diagnóstico suele ser inicialmente clínico, generalmente ante la aparición de fracturas como consecuencia de ligeros traumatismos, cuando existen antecedentes familiares o las fracturas ocurren durante el parto. Durante el embarazo es posible el diagnóstico a través de técnicas de ultrasonidos, como la ecografía, donde pueden detectarse fracturas o deformidades óseas. No obstante, esta forma de diagnóstico es insuficiente y especialmente complicada en los casos más leves.
El estudio genético del ADN es actualmente la única forma de llegar a un diagnóstico definitivo. Es además un análisis cada vez más relevante, siendo probable que en un futuro los tratamientos sean personalizados y estén vinculados a cada mutación específica.
Actualmente la OI no tiene cura, pero existen tratamientos dirigidos a los síntomas que mejoran la calidad de vida de las personas afectadas. Por ejemplo, permiten evitar fracturas y una mejor recuperación de las mismas, disminuyen el dolor o ayudan a mejorar la movilidad. La intervención debe ser siempre multidisciplinar, y en ella intervienen profesionales como pediatras, endocrinos, especialistas en rehabilitación, cirujanos, dentistas, genetistas, fisioterapeutas, psicólogos, trabajadores sociales y terapeutas ocupacionales. No obstante, los principales pilares del tratamiento en la OI son :
Tratamiento farmacológico:
Los bifosfonatos, como el pamidronato o el zolendronato, son el tratamiento farmacológico mas utilizado en la OI. En resumen, inhiben la función de los osteoclastos, células encargadas de la destrucción de hueso en el remodelado óseo, y que en la patología está alterado. En los adultos han demostrado disminuir la frecuencia de fracturas y mejorar la movilidad. También son utilizados en población infantil, donde se ha observado que disminuyen el dolor óseo, mejoran la movilidad y la fuerza muscular, aumentan la masa y densidad ósea y disminuyen el riesgo de fracturas.
Otros tratamientos, aun en fase experimental, incluyen el Denosumab y el anticuerpo esclerostina. Hasta ahora, ambos están demostrando ser eficaces promoviendo el aumento de masa ósea y reduciendo la frecuencia de fracturas.
Tratamiento de fisioterapia:
La principal función de la fisioterapia es maximizar la funcionalidad y movilidad y prevenir la aparición de fracturas a través del fortalecimiento muscular. Así mismo, cumple labores de formación y educación en la movilidad, el manejo y manipulación adecuados de niños pequeños, participa en la rehabilitación tras fracturas y cirugías, en el diseño de ejercicios físicos adaptados a cada persona y en el tratamiento del dolor.
Tratamiento quirúrgico:
Las intervenciones quirúrgicas en la OI comprenden principalmente extremidades superiores e inferiores y columna vertebral. La cirugía en extremidades suele llevarse a cabo tras una fractura o ante la presencia de deformidades que pueden dificultar la marcha o facilitar la aparición de futuras lesiones. En estas intervenciones es común el uso de clavos, telescópicos o no, que ayudan a mejorar el rango de movimiento y la congruencia articular a través de la alineación de las extremidades inferiores.
Los problemas de espalda como la escoliosis, cifosis, espondilosis o espondilolistesis pueden requerir, junto a un manejo a través de fisioterapia, ejercicio y uso de material ortopédico, de una intervención quirúrgica.
La patología presenta una elevada heterogeneidad entre los afectados, habiendo importantes diferencias en las manifestaciones clínicas que presenta cada uno de los tipos. Esto ocurre igualmente dentro de cada tipo, pudiendo expresar, miembros de la misma familia y con la misma mutación, diversos grados de severidad.
A nivel pediátrico, en función de la severidad, la OI puede clasificarse en:
OI de afectación leve:
– Hallazgos en ecografía a partir de 20 semanas de gestación:
- Sin fracturas intrauterinas de huesos largos ni arqueamientos
– Postnatal:
- Raramente fracturas durante el parto
- Crecimiento normal o casi normal
- Sin deformidades en huesos largos
- Deambulación independiente preservada, salvo en momentos de fractura
- Mínimas fracturas vertebrales por aplastamiento
- Puntaje Z de densidad mineral ósea de la columna lumbar generalmente > -1,5 (-1,5 a + 1,5)
- Tasa de 1 o menos fracturas al año
- Sin dolor crónico o mínimo, controlado con analgésicos simples
- Asistencia escolar normalizada, sin ausencias por fatiga, dolor o letargo
OI de afectación moderada:
– Hallazgos en ecografía a partir de 20 semanas de gestación:
- Raramente fracturas intrauterinas de huesos largos o arqueamientos (aunque pueden aumentar en el último trimestre)
– Postnatal:
- Ocasionalmente fracturas durante el parto
- Velocidad de crecimiento y altura afectadas
- Arqueamiento anterior de piernas y muslos
- Arqueamiento de huesos largos debido a inmovilizaciones por fracturas recurrentes
- Aplastamientos vertebrales
- Puntaje Z de densidad mineral ósea de la columna lumbar generalmente > -2,5 y < -1,5
- Tasa prepuberal de más de 1 fractura al año
- Frecuentemente, al menos 5 días de baja escolar por dolor al año
OI de afectación severa
– Hallazgos en ecografía a partir de 20 semanas de gestación:
- Acortamiento de huesos largos
- Fracturas y/o arqueamiento de huesos largos
- Costillas delgadas, con fracturas ausentes o discontinuas
- Mineralización ósea disminuida
– Postnatal:
- Evidente afectación del crecimiento
- Dependiente de silla de ruedas
- Deformidad progresiva de huesos largos y columna vertebral (no debido a fracturas)
- Múltiples aplastamientos vertebrales
- Puntaje Z de densidad mineral ósea de la columna lumbar generalmente < -3
- Tasa prepuberal de más de 3 fracturas al año
- Dolor óseo crónico a menos que se trate con bifosfonatos
- Ausencias escolares habituales debido a fracturas o dolor
OI de afectación extremadamente severa:
– Hallazgos en ecografía a partir de 20 semanas de gestación:
- Acortamiento de los huesos largos
- Fracturas y/o arqueamientos de huesos largos
- Costillas gruesas con reborde continuo debido a múltiples puntos de fractura o costillas delgadas
- Mineralización ósea disminuida
– Postnatal:
- Muslos sostenidos en abducción fija y rotación externa con limitación del movimiento de la mayoría de articulaciones
- Indicadores clínicos de dolor crónico severo (palidez, sudoración, gemidos o muecas en el movimiento pasivo)
- Disminución de la osificación del cráneo, fracturas múltiples de huesos largos y costillas. Tórax pequeño.
- Fémures compactados acortados con apariencia de concertina
- Todas las vértebras aplastadas
- Dificultad respiratoria que lleva a la muerte perinatal
- Curso perinatal letal
Antecedentes:
En general, tenemos 2 copias de cada gen, una copia heredada de la madre y otra del padre. En ocasiones la enfermedad aparece como consecuencia de la alteración de una de las 2 copias, siendo esta una condición “dominante“. Otras veces la patología aparece como consecuencia de alteraciones en ambos genes, tratándose entonces de una condición “recesiva“. A estas alteraciones se les conoce como “mutaciones”. La OI puede ocurrir como consecuencia de ambos patrones.
Existen 4 escenarios en los que podría nacer un niño con OI. Aproximadamente el 60% de los nuevos casos se debe a un patrón de herencia dominante, donde el niño hereda el gen mutado de uno de los padres afectados. En aproximadamente el 20-30% de los casos, la mutación se produce de forma espontanea, no siendo ninguno de los padres portadores de genes mutados. A estos casos se les conoce como “mutaciones de novo“. La herencia recesiva representa alrededor del 5% de los casos de OI, donde el niño hereda dos genes mutados de ambos padres (estos pueden no estar afectados y ser solo portadores, ya que se trata de un gen recesivo). Por último, el mosaicismo es un caso menos frecuente que se explicara a continuación.
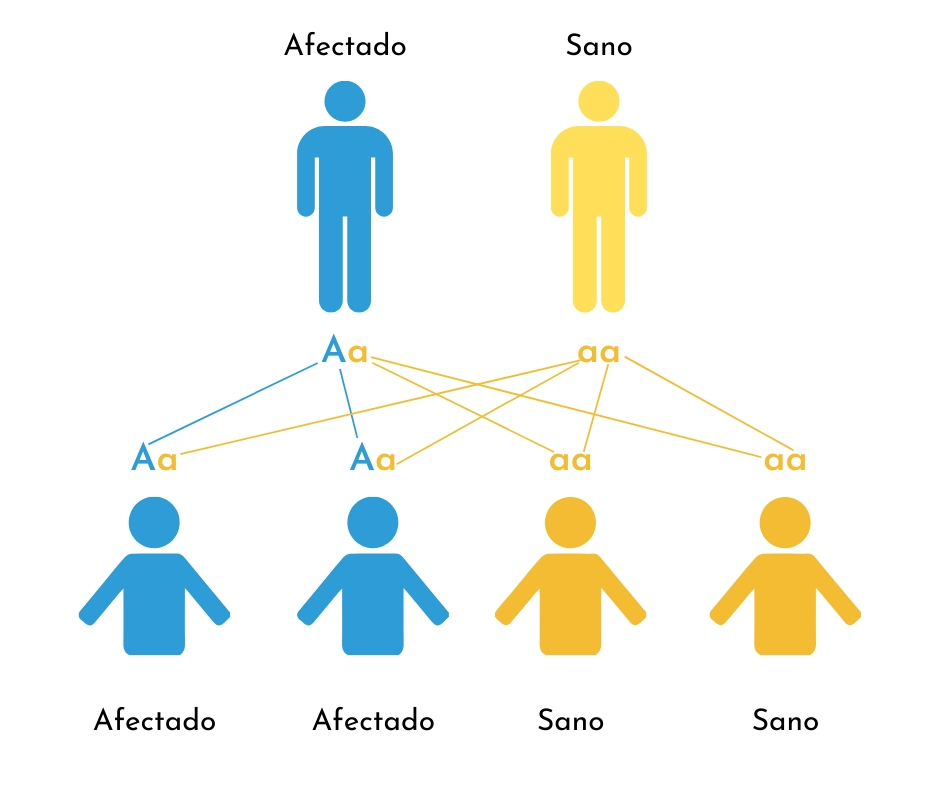
1. Mutación dominante heredada de un progenitor afectado:
Una persona con OI de tipo dominante tiene una copia del gen mutado para el colágeno tipo I, y una segunda copia normal de ese gen. La presencia de una sola copia alterada ya es suficiente para causar OI. Cada vez que la persona afectada concibe un hijo le transmite una de las dos copias, la mutada o la sana, pero no puede decidir cual. Por ese motivo las probabilidades de que el niño nazca con OI son del 50%. La gravedad de la OI en el niño (nº de fracturas, movilidad, estatura, etc.) puede no ser idéntica a la de sus padres. Si el progenitor transmite el gen sano, su hijo no tendrá OI y no podrá transmitir el trastorno a sus propios hijos.
2. Nueva mutación dominante o mutación “de novo”
Este es el caso de la mayoría de niños que nacen con OI de padres sanos y en familias sin antecedentes de la patología. La mutación del gen ocurre antes de la concepción, ya sea en el espermatozoide o en el óvulo especifico que contribuye al embarazo. Este hecho sucede durante la copia normal de genes cada vez que la célula se divide, y no hay un desencadenante ambiental, dietético o conductual conocido que la cause.
En estos casos, los padres no tienen mayor riesgo que la población en general de tener un segundo hijo con OI. El niño afectado que tiene el nuevo gen dominante tiene un 50% de posibilidades de transmitir el trastorno a cada uno de sus hijos. Además, los hermanos no afectados de una persona cuya OI es causada por una mutación espontánea tienen el mismo riesgo de tener un hijo con OI que la población general.

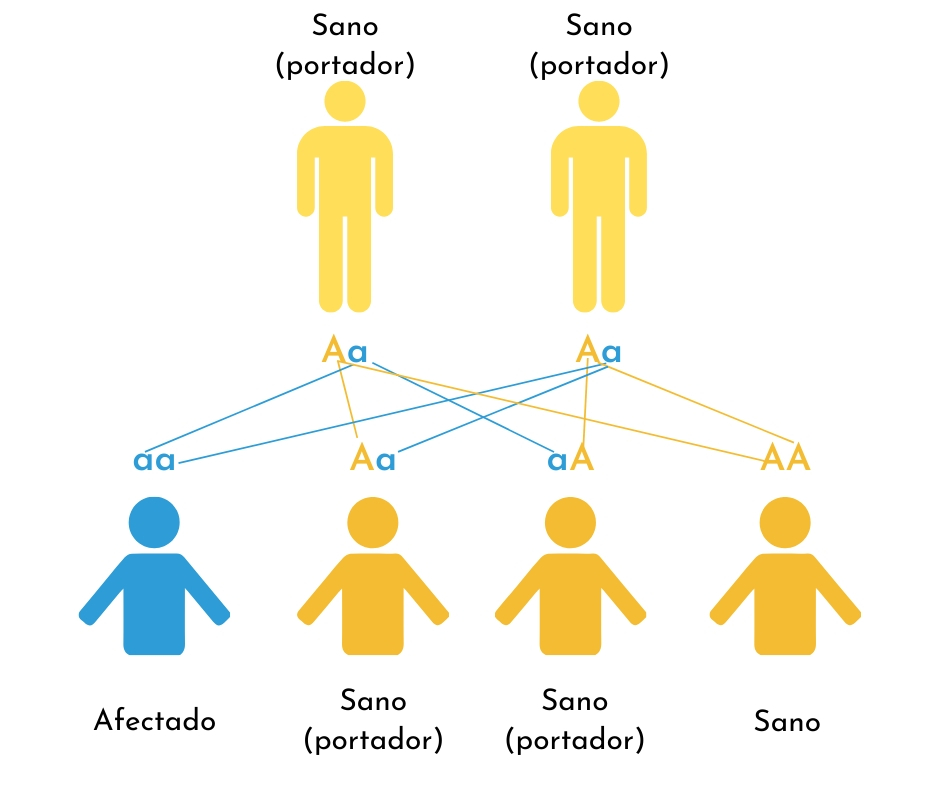
3. Herencia recesiva
La herencia recesiva es una posibilidad a considerar cuando los padres no afectados tienen más de un hijo con OI. En la OI recesiva, ambas copias de un gen en particular recibidas por el niño están alteradas (tienen una mutación). Cada padre del niño afectado es portador de una copia del gen alterado, pero ninguno de los padres está afectado.
En estos casos, existe un 25% de probabilidad de que cada niño reciba una copia alterada del gen de ambos padres y se vea afectado. En promedio, hay un 50% de probabilidad de que el niño reciba una copia normal del gen de uno de los padres y una copia alterada del otro, y que, al igual que el padre, sea portador pero no afectado. Existe un 25% de probabilidad de que un niño herede ambas copias normales del gen. Si un hijo portador tiene una pareja que también es portadora (esto puede ocurrir en los casos en que se prefieren los matrimonios dentro de familias numerosas), la pareja puede tener hijos que estén afectados por una OI grave.
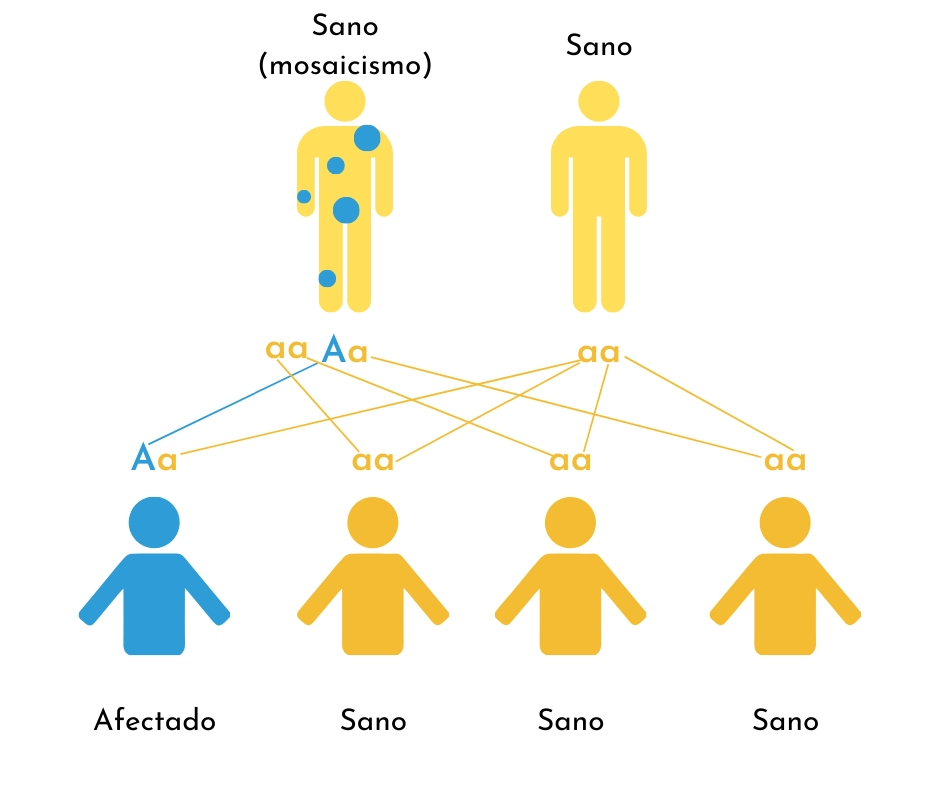
4. Mosaicismo
Hay un pequeño grupo de personas en las que conviven 2 tipos de células, unas afectadas por la mutación genética y otras no afectadas. En ocasiones, estas mutaciones están presentes en las células encargadas de producir óvulos o esperma (mosaicismo gonadal). En estos casos, la persona con mosaicismo no presenta OI, pero tiene probabilidades de transmitir la patología a sus hijos en una forma de “herencia dominante”.
Fuente: www.oif.org Puedes leer el art´culo original en inglés en: https://oif.org/wp-content/uploads/2019/08/Fact_Sheet_Inheritance_Patterns.pdf